Fenylketonurie - klasické příznaky, jako zděděná a dietní terapie
Obsah
- ,1Jak se ukazuje, je fenylketonurie onemocnění
- 2Mechanismus vývoje onemocnění
- 3Fenylketonurie u dětí
- 4Příznaky onemocnění
- 5Příčiny a provokativní faktory
- 6Diagnostika
- 7Léčba klasické fenylketonurie
- 8Výživa novorozenců a dieta
- 9strava pro děti předškolního věku a školní děti
- 10Skupiny produktů v PKU
- 11 , jak kontrolovat hladinu fenylalaninu v krvi
- 12Videa
, výskyt onemocnění je spojeno s genetickými defekty v buněčné stroje - fenylketonurii - je seznam několika dědičných chorob k léčbě. Pioneer tohoto onemocnění byl lékařem z Norska, IA Fellynh později zjištěno, že rozvoj onemocnění a pouze odpovědné genu, tzv fenilalaninhidroksylazy genom (chromozóm dlouhé rameno 12oy obsahující až 4,5% z celkového materiálu buněčné DNA). Dědičná defekt vede k částečné nebo úplné inaktivaci enzymu fenylalanin jater-4-hydroxylázy.
Jak se manifestuje fenylketonurie
fenylketonurie dědičné onemocnění (PKU), vede k chronické otravě organismus toxické látky, které vznikají v důsledku poruchou metabolismu aminokyselin a proceshydroxylací fenylalaninu. Trvalá intoxikace způsobuje poškození centrálního nervového systému (CNS), jehož projevem je postupné snížení inteligence (fenylpyruvicová oligofrenie).
Fellingova nemoc se projevuje v nadměrné akumulaci fenylalaninu v těle a produktech jeho nevhodného metabolismu. Další faktory vývoje fenylketonurie zahrnují přerušený transport aminokyselin přes hematoencefalickou bariéru, nízké hladiny neurotransmiterů (serotonin, histamin, dopamin). Při absenci včasné léčby onemocnění vede k mentální retardaci a může způsobit smrt dítěte.

Mechanismus vývoje choroby
Příčinným faktorem výskytu genových poruch je metabolický blok, který narušuje tvorbu fenylalanin-4-hydroxylázy (enzymu zodpovědného za konverzi aminokyseliny fenylanin na tyrosin). Proteinogenní aminokyselinový tyrosin je nedílnou součástí proteinu a pigmentu melaninu, proto je nezbytným prvkem pro fungování všech systémů těla a jeho nedostatek vede k fermentopatii.
Inhibice metabolitu způsobená mutační inaktivací enzymu je aktivace pomocných cest pro výměnu fenylalaninu. Aromatická alfa-aminokyselina v důsledku vadných procesů výměny se rozkládá na toxické deriváty, které se za normálních podmínek nevytvářejí:
- kyselina fenylpyruvátová (fenylpyruvát) - mastná aromatická alfa-ketoacid, její vzdělánívede k myelinizaci procesů neuronů a demencí;
- kyselina fenylmachtová - produkt vzniklý během regenerace kyseliny fenyl-virionové;
- Fenylethylamin - počáteční sloučenina pro biologicky aktivní vysílače elektrochemických impulzů, zvyšuje koncentraci dopaminu, adrenalinu a norepinefrinu;
- Orthofenylacetát - jedovatá látka, která způsobuje porušení metabolických procesů fekálních sloučenin v mozku.
Lékařské statistiky ukazují, že patologicky pozměněný gen je přítomen ve 2% populace, ale nevykazuje se. Genetická vada se přenáší na dítě od rodičů pouze za přítomnosti onemocnění u obou partnerů, přičemž dítě se v 50% případů stane nosičem mutovaného genu, zůstává zdravým. Pravděpodobnost, že fenylketonurie u novorozenců způsobí onemocnění, je 25%.
Jakým typem je zděděno
Fellingova nemoc je genetická odchylka imitovaná autosomálně recesivním typem. Tento typ dědičnosti znamená, že výskyt příznaků vrozené nemoci se vyskytuje pouze tehdy, když je jedno dítě zděděno z vadné genetické kopie obou rodičů, kteří jsou heterozygotními nosiči změněného genu.
Vývoj vrozené nemoci v 99% případů způsobuje mutaci genu zodpovědného za kódování enzymů, který zajišťuje syntézu fenylalanin-4-hydroxylázy (klasická fenylketonurie). Až 1% genetických onemocnění je spojeno s mutačními změnami, které se vyskytují u jiných genůnedostatečnost dihydropteridin reduktázy (typ II PKU) nebo tetrahydrobiopterin (PKU typu III).
Fenylketonurie u dětí
Klasická forma onemocnění pohlavních orgánů u dětí se ve většině případů projevuje zvenčí zřetelnými příznaky, po 3-9 měsících života. Novorozenci s vadným genem vypadají zdravě, charakteristickým znakem je specifický zvyk (vzhled) dítěte. Symptom se objeví 6-12 měsíců po narození.
PKU typ II se vyznačuje skutečností, že první klinické příznaky se objevují po 1,5 roku od okamžiku nástupu světla. Příznaky onemocnění nezmizí po diagnostice genetických abnormalit a zahájení dietní terapie. Tento typ vrozené nemoci často vede k smrtelnému výsledku 2-3 let života dítěte. Nejběžnější příznaky typu PKU typu II jsou:
- vyjádřili odchylky v mentálním vývoji;
- hyperreflexie;
- porušení motorických funkcí všech končetin;
- syndrom nekontrolovaných svalových kontrakcí.
Klinické příznaky mutačních změn v genu typu III jsou podobné onemocněním vyskytujícím se u typu II. Nedostatek tetrahydrobiopterinu je charakterizován triádou specifických příznaků:
- vysoký stupeň mentální retardace;
- zdánlivě snížená velikost lebky vzhledem k ostatním částem těla;
- spasticita svalů (s možností úplné ztráty pohybu končetin).

projevy Fellingovy nemoci
V průběhu klinických studií a pozorování bylo navrženo, že tento účinek byltoxický výměna deriváty fenylalanynovoho způsobuje pokles mentálních schopností, který je progresivní a může vést k demenci (mentální retardace, hloupost). Mezi možné příčiny ireverzibilní poruchy mozkové činnosti považuje za přiměřené vzhledem k nižším úrovním tyrosin nedostatku neurotransmiterů, které přenášejí impulsy mezi neurony.
Přesná příčinná souvislost mezi dědičné onemocnění a mozkové poruchy dosud byly identifikovány jako mechanismu fenylketonurií kvůli psychických stavů, jako je эhopraksyya, echolalia, záchvaty hněvu a podrážděnost. Výsledky těchto testů ukazují, že fenylalanin má přímý toxický účinek na mozku, který může také způsobit snížení inteligence.
Struktura a fenotypové zvláštnosti
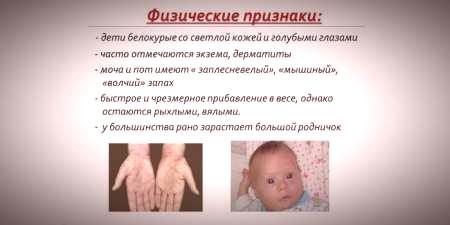
Vzhledem k tomu, že se nasycení pokožky a vlasů pigmentu závisí na úrovni tyrosinu v mitochondriích hepatocytů a způsobuje fenylketonurií zastavení přeměnu fenylalaninu, u pacientů s tímto onemocněním mají fenotypové vlastnosti (recesivní). Zvýšený svalový tonus způsobuje výskyt odchylek v struktuře těla - stává se dysplastickým. Vynikající vnější vlastnosti fenylketonurie zahrnují:
- hypopigmentace - lehká kůže, světle modré oči, změkčené vlasy;
- cynismus končetin;
- snížená velikost hlavy;
- zvláštní pozici - když se snaží stát nebo sedět dítě vtělí „na míru“ (ruce a nohy ohnuté v kloubech).
Příznakynemoci
Fellingova nemoc se včas úspěšně léčí přizpůsobením výživy a rozvoj dítěte v závislosti na věkové skupině. Obtížnost v detekci genové mutace spočívá ve skutečnosti, že počáteční příznaky jsou obtížné odhalit i zkušený pediatr. Závažnost příznaků vrozeného onemocnění se zvyšuje s rostoucím věkem dítěte, protože použití proteinových potravin přispívá k rozvoji poruch CNS.
Známky u novorozenců
Během prvních dnů života dítěte je obtížné zjistit příznaky patologických abnormalit - dítě se chová přirozeně, nedochází k žádnému zpoždění ve vývoji. Příznaky onemocnění se nejdříve objevují po 2-6 měsících po porodu. Rodiče by měli být obezřetní vůči chování dítěte, které je charakterizováno nízkou aktivitou, letargií nebo naopak úzkostí, hyperbolektivitou.
Při nástupu kojení začínají nové bílkoviny vstoupit do těla novorozence mlékem, což slouží jako katalyzátor výskytu prvních příznaků, což jasně ukazuje, že onemocnění začalo pokračovat. Specifické klinické projevy onemocnění zahrnují:
- trvalé zvracení (často užívané pro vrozené zúžení brankáře);
- častá dislokace;
- žádná reakce na vnější podněty;
- svalová dystonie (snížení svalového napětí);
- konvulzivní syndrom (záchvaty epileptického nebo neepileptického typu).
Symptomy u dětí po 6 měsících
Pokud projev genetického onemocnění není(nebo nebylo vidět) během prvních 6 měsíců od narození dítěte, pak po tomto období již můžete přesně určit zpoždění v psychomotorickém vývoji. Symptomy genetických poruch způsobených deficitem enzymu u dětí starších šesti měsíců jsou:
- snížení aktivity (až do úplné lhostejnosti);
- nedostatek pokusů vstát, sedět;
- zvláštní "myší" vůně kůže (vůně plísní vznikají v důsledku stažení toxických derivátů fenylalaninu prostřednictvím potních žláz a moči);
- ztráta schopnosti vizualizovat tváře rodičů;
- olupování kůže;
- výskyt dermatitidy, ekzému, sklerodermie.
Progrese nemoci při absenci léčby v dětství
Pokud vývojové odchylky nebyly zjištěny v dětství a nebyla provedena vhodná léčba, onemocnění se aktivně rozvíjí a často vede k postižení. Nedostatečná terapie v počátečním stádiu onemocnění způsobuje výskyt takových symptomů onemocnění ve věku 1,5 roku:
- mikrocefalie (snížená velikost mozku);
- odtok (posunutí horní řady zubů dopředu);
- pozdější erupce zubů;
- hypoplázie skloviny (tenká nebo úplná absence zubní skloviny);
- zpoždění jazykového vývoje až do úplného nedostatku řeči;
- 3, 4 stupeň oligofrenie (mentální retardace, mentální retardace);
- vrozené srdeční onemocnění (defekty ve struktuře srdečního svalu, srdce,velké plavidla);
- poruchy vegetativního systému (akrocyanóza, zvýšené pocení, arteriální hypotenze);
- zácpa.
Příčiny a provokativní faktory
K prokázání mutace s autosomálně recesivní dědičností musí být z obou rodičů zděděn defektní gen. Genetické onemocnění tohoto typu se vyskytují na stejné frekvenci u novorozených chlapců a dívek. Patogeneze PKU je způsobena narušením výměny fenylalaninu, která se může vyskytnout ve třech formách. Léčba dietní terapií podléhá klasické fenylketonurii typu I.
Atypické formy onemocnění lze vyléčit úpravou výživy. Tato odchylka je způsobena nedostatkem tetrahydropterinu, dehydropterin reduktázy (zřídka - pyruvatetetrahydropterinstanzy, guanosin-5-trifosfát cyklohydrolázy atd.). Většina případů smrtelných případů byla zaznamenána u pacientů s vzácnými variacemi PKU, s klinickými projevy všech podobných onemocnění. Riziko porodu s mutovaným genem fenylalaninhydroxylázy se zvyšuje, pokud jsou rodiče blízkými příbuznými (s úzce souvisejícími sňatky).
Diagnostika
V případě podezření na genetickou poruchu je diagnóza stanovena na základě shromáždění údajů získaných v důsledku studie anamnézy onemocnění - genealogické údaje, výsledky klinického a lékařského genetického výzkumu. Pro včasnou detekci vrozených onemocnění (PKU, cystickou fibrózu, galaktozémii apod.) Program povinné hmotnostiLaboratorní vyšetření všech novorozenců (neonatální screening).
Pokud si budoucí rodiče uvědomují přítomnost mutovaného genu, moderní medicína nabízí způsoby zjištění vady během těhotenství (prenatální diagnostika plodu invazivním způsobem). Pro rozdělení fenylketonurie podle druhu závažnosti je použita podmíněná klasifikace založená na hladině fenylalaninu ve fibrinogenní tekutině získané z plazmatické plazmy:
- Těžká fenylketonurie - 1200 mkmol /l.
- Průměr - 60-1200 μmol /l.
- Snadná (bez nutnosti léčby) - 480 μmol /l.
Screening test
Detekce genetických abnormalit nastává v několika fázích. V první fázi v nemocnici u všech dětí 3-5 dní života se provádí vzorkování periferní krve (z pěti) pro výzkum. Materiál se aplikuje na papírovou formu a odešle do biochemické laboratoře, kde probíhá její biochemická analýza. Ve druhém stupni skríningového testu se stanoví koncentrace fenylalaninu normální hodnoty.

Pokud nejsou zjištěny žádné patologické změny, diagnostika skončí, což je zaznamenáno na kartě dítěte. Při výskytu odchylek od normy se výsledky diagnózy posílají na doktora-pediatra, aby poskytl studium refinementu krevního vzorku novorozence. Zdraví dítěte závisí na včasném a přesném provedení všech opatření k detekci abnormalit. Pokud je diagnóza potvrzena po opakovaném screeningovém testu, budou rodiče dítěteposlaný na kliniku pro genetiku dítěte za účelem léčby.
Analýzy a studie pro potvrzení diagnózy
Opakovaná diagnostika v případě zjištění odchylky od normy během počáteční screeningové zkoušky se provádí opětovným posouzením analýz. Kromě stanovení obsahu fenylalaninu v krvi metodami diagnostiky PKU u dětí a dospělých patří:
- Zkouška tvoření - stanovení kyseliny fenylpyruvicové v moči přidáním chloridu železitého k biomateriálu (dochází k modrozelené barvě);
- Test Gatree - hodnocení míry reakce mikroorganismů na metabolické produkty nebo enzymy obsažené v krvi pacienta;
- chromatografie - studium chemických vlastností látek rozdělených mezi dvě fáze;
- fluorimetrie - ozáření biomateriálu monochromatickým zářením za účelem určení koncentrace látek obsažených v tomto materiálu;
- elektroencefalografie - diagnostika elektrické činnosti mozku;
- zobrazování magnetickou rezonancí - narušení atomových jader buněk elektromagnetickými vlnami a měření jejich odezvy.
Léčba klasické fenylketonurie
Základem léčby fenylketonurie je omezení spotřeby produktů, které jsou zdrojem bílkovin živočišného a rostlinného původu. Jediným způsobem úspěšné léčby je dietní terapie, jejíž přiměřenost je odhadována obsahem fenylalaninu v séru. Maximální přípustná hladina aminokyselin u pacientů různých věkových skupinje:
- u novorozenců a dětí do 3 let - do 242 μmol /l;
- pro předškolní děti - až 360 mkmol /l;
- u pacientů ve věku od 7 do 14 let - až do 480 μmol /l;
- u dospívajících - až 600 μmol /l.
Účinek stravy závisí na tom, v jakém stadiu onemocnění je korekce stravy. S včasnou diagnostiku vrozených vad stravy jmenovaný od 8 týdnů života (po uplynutí této doby jsou nevratné změny začínají). Nedostatek včasné opatření vede ke komplikacím a nižší IQ při 4 ° C po dobu 1 měsíce od narození do začátku léčby.

Vzhledem k tomu, že terapeutická strava pro fenylketonurií poskytuje úplné vyloučení ze stravy živočišných bílkovin, je potřeba dalších zdrojů esenciálních aminokyselin a vitamínů B, vápníku - a fosfor obsahující minerální sloučeniny. Výrobky, které mají být přidány k neproteinové dietě, zahrnují:
- proteinové hydrolyzáty (Amygens, Aminazole, Fibrinoses);
- neobsahují fenylalaninové směsi nasycené esenciálními aminokyselinami - Tetrafen, bez fenylů.
Kromě terapeutických opatření k řešení příčin porušení fungování těla by měla být symptomatická léčba zaměřená na odstranění vad a normalizaci koordinace řeč motoru. Komplexní terapie zahrnuje fyzioterapeutické procedury, masáže, pomoc řečnického terapeuta, psychologa, výkon gymnastických cvičení. V některých případech s dietní terapiíukazuje použití antikonvulzí, nootropních a vaskulárních léků.
Vlastnosti léčby atypických forem
fenylketonurie II a typ III neléčitelné nyzkobelkovoy dieta - hladina fenylalaninu v krvi zůstává konstantní, zatímco pro omezení přenosu proteinu v těle nebo klinických symptomů postupuje i při nižších úrovních aminokyselin. Účinná léčba těchto forem onemocnění se provádí za použití:
- tetrahydrobiopterin - faktor ovlivněného enzymu;
- tetragidrobiopterina syntetické analogy - tyto látky jsou lépe pronikat hematoencefalickou bariérou;
- náhradní terapie léky - neodstraňují příčinu fenylketonurie, ale zachovat normální fungování těla (levodopa spolu s Karbydofoy, oksytryptofan 5, 5 formyltetrahydrofolat);
- hepatoprotektory - podporují fungování jater;
- antikonvulziva;
- zavedení genu fenylalaninhydroxylázy do jater - experimentální metoda.
Funkce výživy novorozenců a dietní terapie
V prvním roce života s PKU přijatelnými kojením, ale jejich počet by měl být omezen. Od 6 měsíců přijatelnou úroveň příjmu fenylalaninu je 60 až 90 mg na 1 kg tělesné hmotnosti dítěte (100 g mléka obsahuje 5,6 mg fenylalaninu). Počínaje 3 měsíci by měla být dieta dítěte postupně rozšiřována zaváděním ovocných šťáv a brambor.
Děti od 6 měsíců umožněn vstup do stravy kaší zeleninu, obiloviny (s sága) bezbilkovyhpolibky Po 7 měsících můžete dávat baby bílkovinné těstoviny s 8 měsíci - bez bílkovin. Věk, kdy je přenos bílkovin v těle nemocného dítěte omezen, není stanoven. Lékaři stále diskutují o účelnosti dietní terapie, ale souhlasí s tím, že by měla být dodržována výživa ve věku minimálně 18 let.
Fenylketonurie, diagnostikována se ženou, není důvodem k ukončení narození dítěte. Těhotné ženy s PKU, aby nedošlo k poškození plodu během těhotenství a je nutné před plánovaným početím a během těhotenství dětské stravy s omezením fenylalaninu prevence možných komplikací (jeho hladina v krvi by měla být až 242 mmol /l).
Vakcína pro kojence
Dieta pro fenylketonurií na základě snížení podstatné dávky přirozené vlákniny v každodenní stravě, ale tělo novorozence může vyvíjet normálně při absenci potřebných mikronutrientů. Pro naplnění potřeb dítěte laktózy použité ve směsi kyseliny protein amino, který, podle ruského práva, musí být k dispozici u pacientů zdarma.
Tělesná tolerance k fenylalaninu během prvního roku života se rychle mění, takže je nutné kontrolovat jeho koncentraci v krvi dítěte a upravit stravu. Směsi jsou určeny pro určité věkové skupiny:
- byl jmenován na děti Afenylak 15. Analog-SP, PKU-1, PKU-mix, PKU Anamix;
- pro děti starší než 1 let, Předepsaných obohacené o vitaminy a minerální látky ve směsi s vysokým obsahem bílkovin - PKU Prima, P-AM Universal, PKU-1, s PKU-2, Maksameyd XP, XP Maksamum.

, dietní výrobky pro doplňování protein
Jedním z hlavních složek dietní jídla s fenylketonurií jsou malobelkovыe produkty na bázi škrobu. Tyto doplňky obsahují hydrolyzát kaseinu, tryptofan, tyrosin, methionin, dusík a poskytují denní potřeby dítěte v proteinu, který je nezbytný pro normální růst a vývoj. Specializované produkty, zaplnit nedostatek esenciálních minerálů a aminokyselin pro jejich nedostatek v potravě jsou:
- Berlofen;
- Tsymorhan;
- Mynafen;
- Aponty.
strava pro děti předškolního věku a školního věku
Vzhledem k přizpůsobení fenylalaninu dětí ve věku od 5 let mohou postupně snižovat dietní omezení. Sací expanze je zavedení obilovin, mléčných výrobků, masných výrobků. Středoškolští studenti již mají vysokou toleranci k fenylalaninu, protože v tomto věku může pokračovat v rozšiřování stravu s ohledem na nutnost sledovat reakci na změny ve stravě. Pro kontrolu dítěte, následujícími způsoby:
- Neurologické hodnotících ukazatelů psychický stav;
- monitorovací ukazatele elektroencefalogram;
- stanovení hladiny fenylalaninu.
skupiny produktu s PKU
ve stravě pacientů s PKU, spolu s malobelkovыmyMezi škrobové produkty a léčebné směsi patří také produkty přírodního původu. Při sestavování menu by měl jasně vypočítat příjem bílkovin a nepřekračujte doporučené dávkování lékař. Pro odstranění toxické účinky na tělo vyvinuty 3 seznam výrobků, který obsahuje zakázané (červená) zastaralý (oranžová) a povolenou polohu (zelená).
Červený seznam
fenylketonurie vyvíjí v nepřítomnosti enzymu, který konvertuje fenylalanin na tyrosin jako vysoký obsah bílkovin je základem pro klasifikaci produktů v seznamu zakázaných (červená). Položky v tomto seznamu by měly být zcela vyloučeny dietu pro pacienty s PKU
- maso;
- vnitřní orgány zvířat, vedlejší produkty;
- klobásy, klobásy;
- mořské plody (včetně ryb);
- vejce všech ptáků;
- kysané mléčné výrobky;
- matice;
- luštěninové a obilné plodiny;
- sójové produkty;
- želatinové nádobí;
- cukrovinky;
- aspartam.
Orange list
Výrobky, které mají dávkování požití dítě je diagnostikována s PKU jsou zahrnuty v seznamu oranžové. Zahrnutí položek z tohoto seznamu do stravy je přípustné, avšak v přísně omezených množstvích. I když tyto produkty neobsahují mnoho bílkovin, ale můžu také zvýšit hladinu fenylalaninu, proto se jejich použití nedoporučuje:
- konzervovaná zelenina;
- bramborové a rýžové pokrmy;
- zelí;
- mléko;
- sherbet.
Zelený seznam
Neproteinové přípravky jsou povoleny pro použití u pacientů s fenylketonuriíí bez omezení. Před zakoupením položek ze zeleného seznamu je třeba zkontrolovat složení uvedené na obalu a ujistit se, že neobsahuje fenylalanin obsahující barvivo aspartamu:
- ovoce;
- zelenina (kromě brambor a zelí);
- bobule;
- zelené;
- škrobové obiloviny (ságo);
- med, cukr, džem;
- výrobky z mouky z kukuřice nebo rýžové mouky;
- máslo, tuky (máslo, slunečnice, olivy).
Jak kontrolovat hladinu fenylalaninu v krvi
Fenylketonurie je nevyléčitelné onemocnění, které může být přeměněno na fázi stagnace pomocí dietní terapie a terapeutických a profylaktických opatření. Při změně životních podmínek, porušení stravy, může být onemocnění opět zhoršena, takže pacienti potřebují celoživotní pozorování. Kontrolním postupem je pravidelně stanovovat hladinu fenylalaninu v krvi. Frekvence podání závisí na věku pacienta:
- až 3 měsíce - vyšetření krve by mělo být provedeno jednou týdně, dokud nebudou získány stabilní výsledky;
- od 3 měsíců do 1 roku - 1-2 krát měsíčně;
- od 1 do 3 let - 1 za 2 měsíce;
- za 3 roky - čtvrtletně.

Krev pro analýzy se zdá být 3-4 hodiny po jídle. Kromě screeningu je vývoj PKU řízen stanovením nutričního stavu, fyzickým, emocionálním vývojem pacienta, úrovní intelektuálních schopnostía rozvoj jazyka. Výsledky pozorování může potřebovat další diagnózu účasti příslušných odborníků.
Video
Informace uvedené v článku jsou informativní. Materiály tohoto článku nevyžadují nezávislou léčbu. Pouze kvalifikovaný lékař může diagnostikovat a poradit s léčbou na základě individuálních charakteristik konkrétního pacienta.













